# 热门搜索 #
大模型
人工智能
openai
融资
chatGPT
大家可能从小就听说过爱迪生尝试上千种材料作为灯丝,凭着不断试错方法以及永不言弃的精神,最后研发出日用白炽灯的故事——天才靠的是百分之一的灵感和百分之九十九的汗水。
然而,随着科学的进步和现代社会的发展,新型材料的研发变得愈发复杂。现在,研究者经常需要在上百万大小的材料空间同时优化数十个不同的性质,以寻找适用于电池、半导体、催化剂和合金等领域的新材料。如果说爱迪生需要 99% 的汗水,那现在研究者可能需要 99.99% 的汗水。
而新型材料的研发历程就有点像人们去「沙滩」上捡「贝壳」,在「沙滩」表面好找的贝壳在大家不断发掘下已经被拾的差不多了,更漂亮的「贝壳」却还埋在在更深的「沙滩」下面。这些「贝壳」可能用传统的方法很难发掘,但 AI for Science(简称 AI4S)为我们带来了新的可能。
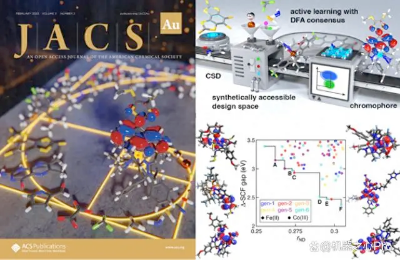
Microsoft Azure Quantum 研究科学家、麻省理工学院(MIT)段辰儒博士在吸光团簇的研发方面遇到了和「爱迪生类似的难题」——寻找吸收长波可见光并且激发态稳定的过渡金属吸光团簇。吸光团簇的发现在现阶段有两大难点,首先其设计空间有三千万个分子,比爱迪生当年大的多。此外,长波可见光吸收与稳定激发态是化学上接近互斥的两个性质,增加了探索难度。
段博士首先使用了「爱迪生式」的随机采样,尝试了两千个分子,发现无一同时满足这两种性质。继续类似探索带来的希望非常有限。于是,段博士带着 AI4S 的角度重新审视了这个问题:在这两千个分子的数据下建立了机器学习模型来快速预测分子的吸收波长和激发态时长;同时,使用主动学习与贝叶斯优化引导探索这硕大材料空间。在新的探索方式之下,模型找到目标分子的概率随着探索过程的进行迅速增长。达到在五百个分子后,每尝试五个分子,就可以找到一个吸收长波可见光并且激发态时间长的过渡金属吸光团簇。保守估计,这种方法的改进带来了将近 1000 倍的加速,相当于将原本三年的研发周期缩短到了一天!段博士的工作也于近期作为封面文章发表在 JACS Au。
论文链接:Duan et al.,
https://doi.org/10.1021/jacsau.2c00547
在 AI 快速发展的时代。作为个人,应该怎么样把这些技术与我们领域知识结合在一起?AI4S 当前研究进展处于什么阶段?AI4S 前景如何?我们一起看看专注于「AI+化学/材料」领域的研究者贾皓钧博士和段辰儒博士,怎么看待 AI4S。以及两位在 AI4S 领域的相关探索研究。
贾皓钧(左)和段辰儒(右)
ScienceAI:两位能介绍一下自己吗?
段辰儒:我目前在 Microsoft Azure Quantum 做研究科学家,主要研究生成式 AI 和大模型在化学方面的应用,和微软 AI4S 研究的产品化。两年前在 MIT 获得化学博士学位,博士期间主要做 AI4Chemistry 的研究,开创了 AI 决策模型在高通量计算中的整合,使得 AI+计算更好的服务于化学和材料发现。工作之余,我也一直参与组织在机器学习会议上的 AI4S workshop(
https://ai4sciencecommunity.github.io/neurips23.html) 系列,推动社区发展。
贾皓钧:我目前在 MIT 化学系和化工系博士第五年,博士期间师从 AI 助力化学设计领军人物 Heather Kulik,主要研究方向是结合高通量量子化学和人工智能来帮助发现用于碳中和的新型催化材料。我之前也曾在陶氏化学核心研发部门进行研发工作,致力于使用 AI 来研发催化剂配方以及预测化学反应的路径。除了科研方面,我之前也担任过麻省理工学院中国学生学者联合会(MIT CSSA)2022-2023 主席。
ScienceAI:我们了解到两位之前本科都毕业于物理系,能给我们讲一下从本科到现在的这段科研和工作经历吗?因何与 AI 结缘的?
贾皓钧:我本科是学物理的,在上学期间研究比较广泛,主要做过二维材料,电池,超导,合金的第一性原理计算模拟。而那时 PyTorch 和 TensorFlow 还没流行,但大量的数据也让我意识到材料筛选的重要性,这也因此打下了我与 AI 结缘的伏笔。后来到美国之后,发现结合 AI 算法可以大规模的来进行高通量筛选和预测。之前几个模拟等很多天出的结果,现在结合 AI 算法可以进行million 材料数量级别的高通量筛选和预测。从我迈入科研的大门到如今短短十年时间,计算模拟已经不再是单纯的验证实验,而是真实应用到工业界。AI4S 的范式是一个真实在发生的事,有很大的市场潜力。
至于为什么转向偏化学,是因为后来我发现物理离现实稍微有点远。我当时做学术的一个大的目标是想「做出的成果既能上得了书架,也能上得了货架」。我更想做一些偏实际的东西,所以本科也在约翰霍普金斯大学化学系待过,做一些气相表面化学的实验研究。其实那个时候是第一次接触化学,但发现了很多有意思的地方。例如,物理多数情况关注现象本身,即宇宙本身的规律。化学更多关注的是过程和改变,是我们可以进行人为调控的。在这之中,我们能找到一些真正有价值的东西。这也是我从物理专业转向化学的原因。
段辰儒:我跟皓钧的经历比较相像,我本科做的是理论凝聚态,研究有趣的量子相变等现象。尽管这些研究领域引人入胜,但与实际应用之间存在一定距离。在这过程中,我慢慢意识到相比于探索世界底层的物理规律,我更加享受使用物理规律改造这个世界。于是在攻读博士学位时,我渴望朝着更接近应用的方向发展。来到 MIT 之后也跟很多老师交谈,其中与我导师聊天时,有一个点非常吸引我,她提到组里一名学生一篇文章产生的数据量比她来 MIT 之前整整 8 年的研究总和还多。这让我在 2017 年就意识到了计算化学领域的发展速度迅猛。
我开始阅读与研究相关的几篇论文,并发现尽管计算化学的计算能力和通量不断增加,但整个计算流程,即如何整合各种计算,尚未得到充分的发展。因此,我猜想将 AI 融入高通量计算体系,构建更优秀的工作流会是学术方面的一个增长点。2017 年,我在 MIT 开始自己的博士生涯。虽然当时还没有 AI4S 的概念,但我已经认识到高通量计算和人工智能相结合的潜力巨大,以及它可能对各学科带来范式的变革。于是我就坚定的选择了这个领域,实现了从物理到化学及工程应用方向的转变。
ScienceAI:两位博士目前都专注于「AI+化学/材料」计算领域的研究,能给我们讲一下 AI 如何助力计算化学吗
贾皓钧:量子化学是通过第一性原理计算模拟材料的性质,具体的思路是:先固定原子位置解薛定谔方程,得到材料的电子结构,然后基于电子结构获得这个材料的性质,例如:电子能量、力学性质和结构性质等等。第一步解薛定谔方程是计算量极大的量子多体问题,它的计算量随着模拟的原子数指数增加,也是目前最难解决的,甚至可以说是从头算模拟领域的「卡脖子」问题。薛定谔方程的复杂性使其难以求解。但随着 AI 技术的发展,我们可以通过元素的內禀属性(原子半径、电负性、价电子等等)、材料的键长键角等等,「绕过」薛定谔方程多体问题,直接预测材料的性质。有两种主要思路:
通过 AI 预测电子结构来获取材料性质:这一方法的优点在于其物理基础,即从电子结构到性质的转化是清晰的,没有黑箱。然而,挑战在于处理材料的波函数,它涉及到大量的矩阵,并且受到电子数量的限制,这限制了模拟的规模。AI预测波函数也很具挑战性。一种可行的方法是将材料的电子波函数投影到原子基组上,然后通过 AI 预测投影系数。然而,这会引发物理和数学上的问题,如基组的完备性等,这些问题在将其应用于实际系统时会带来技术挑战。
通过 AI 预测材料性质而不考虑电子结构:这种方法的优势在于完全规避了多体问题,理论上可以用于处理大型体系。然而,这种方法高度依赖于大量的训练数据。相较之下,第一种方法可能有助于发现新的物理机制。不过,第二种方法已广泛用于新材料的探索和设计,显著加速了材料研发周期。例如,结合高通量计算与 AI 来预测性质,可以快速筛选出受关注的催化剂、电池、二维材料等。AI也能用于预测原子受力,加速分子动力学模拟或结构搜索,从而提高材料模拟的规模和精度。
段辰儒:皓钧已经说了很多量子化学怎么样来帮助我们来算一些材料的性质。但其实还有一个问题:这么多量子化学方法,都会涉及到一些近似,那选取不同的近似就会给出不一样的结果。那怎么样来用 AI 帮助我们从怎么多计算方法中选择一个「最合适」的?或者怎么样能够利用多个「近似的结果」得到一个「更加准确的结果」?最近受抖音「用户—短视频」匹配逻辑的启发,我想到可以做「化学材料—计算方法」匹配。通过搭建「密度泛函推荐器」,我们首次将金属有机配合物的高通量计算的准确度逼近了实验测量的误差精度。这个文章也发表在了 Nature 大子刊 Nature Computational Science 上,并获得了 Nature 新闻评论的关注。我认为这些把 AI 决策模型融合进计算流程的想法是「抹平」计算和实验结果差距的关键。
ScienceAI:传统的化学/材料研究方式是什么样的?有没有比较成熟的方法理论,材料数据库或者平台?
贾皓钧:传统的材料研究方法是试错法,就像爱迪生试验灯丝一样,比如逐一替换元素。以前一段特别火的超导为例,La-Ba-Cu-O 超导体是最早的铜基超导体,获得了诺贝尔物理学奖,但是它只有 35 K 的超导,低于液氮温区,但是 Y替换 La 之后,Y-Ba-Cu-O 超导体的超导温度高于液氮温区,使得铜基超导体被广泛引用。传统的研究方式就特别像以前的「手工作坊」,材料研发周期非常长,而且耗费的人力物力也是巨大的,并且存在偶然性。
随着计算机技术和量子力学理论的发展,基于密度泛函理论的材料预测方法变得成熟。结合结构搜索算法和高通量计算,我们可以更有效地筛选候选材料,从而节约试错成本。从计算角度,多种元素排列组合和 230 种空间群,造成材料的相空间是极大的,在不改变第一性原理算的基础上,预先筛选材料是比较成熟的算法,如 CALYPSO、USPEX 和 AIRSS 等。近几年超导材料研究的活跃与氢基超导体的发现有关,H3S 在 150 GPa 下表现出了 203 K 的超导转变温度。然而这个重要的发现是通过遗传算法指导,在高压实验获得的。此外,元素替代和密度泛函计算结合数据库也是一种有效的路径。著名的数据库有 material project,material cloud 等等。然而,传统方法仍受到多体问题的限制,计算成本仍然昂贵,而 AI 的出现提供了一种可能解决这个问题的途径,从根本上解决了多体问题,使材料研究变得更高效。
段辰儒:我们科学探索的方式目前还是偏试错主导的。经常是一些偶然的不可控因素带来了一大波新发现。其实整个 AI4S 的核心在于科学发现的体系化。比如当我们产生了大量数据的时候,我们可以用这些数据来建 AI 模型,之后更加定向的探索这个空间,而不是去随机的探索。以及我们可以用一些生成式 AI,不去筛选整个空间,反而来生成一些新的分子和材料。这些方法论上的改变,最终会带来范式的迁移,使整个科学研究更加体系化,平民化。这样,能够让更多的人以更加低的壁垒参与到科学研究当中,加快科学发现的迭代,提高科学研究的成果转化率。
ScienceAI:AI4S 会使这种科学发展的「意外之喜」(偶然性)越来越少吗?
段辰儒:趋势是「意外之喜」会越来越少,「意料之喜」会越来越多,总体而言「喜大于忧」。在任何的优化中,行为模式都可以分为两类,一个的是利用已有的先验知识,来进行下一步的选择。比如,皓钧刚说的超导体中元素的同族元素替代。第二个是主动探索新的未曾探索过的材料体系,LK-99 的这类的尝试。这其实和我们去餐馆点菜很像。先验知识就像我们对吃过的菜的评价和印象,主动探索就像我们有时心血来潮点了商家的新品推荐。
优化理论中贝叶斯优化有很多讨论如何平衡这两种互补的行为来最大化探索的收益。那 AI4S 其实就是通过不断的改进,通过科研工具库里面的工具来调整这些平衡,导致我们最后的科研产出或者收益最大化,我们开头介绍的 「寻找吸收长波可见光并且激发态稳定的过渡金属吸光团簇」的工作就是一个典型的例子。它相当于你以「试错法」在饭店吃了 100 个菜,都又贵又难吃,在你快把这家餐厅拉黑的时候,发现 AI4S 的优化方法可以精准帮你点到这家餐厅便宜又好吃的菜。
另外,随着生成式 AI 和扩散生成模型的发展,那我们的科研工具库里甚至有了主动生成意外的可能性。之前的研究都是在一个大的材料库里去筛材料,这其实相当于自己限制了自己的想象空间。那生成式 AI 就会带来一些「意外之喜」,因为生成的材料很可能就不在原有的材料库里。这相当于饭店里的菜难吃不要紧,咱可以直接用 AI 生成新的菜单!这些模型其实已经被应用到了很多科学研究中,比如生成一些能量比较稳定的分子,以及对于一个蛋白质和靶点生成小分子药物等等。
最近,我们做一个项目使生成式 AI 不拘泥于生成一个单一的结构,而是直接生成化学这门学科研究的核心—全新的化学反应。我们戏称这个方法为现代「AI炼金术」哈哈,因为它背后的原理虽然有统计学理论的支撑,但在应用层面就像古代炼金术一样可以帮助我们直接探索完全未知的化学反应,生成符合统计分布的「意料之喜」。
ScienceAI:ChatGPT 已火爆全球,之后出来很多基础科研领域的「类 ChatGPT」,你们觉得当前科研领域的大模型研究处于什么阶段?未来会有咋样的一个「科研 ChatGPT」?
段辰儒:之前有一本很火的书《Quantum Physics for Babies》(宝宝的量子物理学)。我觉得现在的状态就是 「Science GPT for Babies」。主要原因是 ChatGPT 是 2022 年底出来的,时间还很短,大家的惯性思考仍然停留在怎么样把 GPT 直接应用在原本的问题上。这导致目前在 Science 方面,大部分工作都是一些简单的GPT直接应用,或者做一些微调。但这个阶段很快就会过去。GPT 本身真正带来的潜力,是改变了人们与机器,以及不同的机器之间相互交互的方式。比如原本需要会写 Python,我才能做一些机器学习的东西。那我现在很多写代码的任务可以交给 GPT。或者为了做机器人,我可能需要会写 VB 这样的「古老代码」,因为那些芯片都是老的。那我现在可能只用自然语言也可以做这个交互。
未来,我觉得值得探索的方向是以 GPT 为核心,串联复杂的研发过程。以及在工业生产链条中,把它作为自然语言的接口,这样就可以降低人们学习各种复杂的软件、其他编程语言以及各种仪器之间的壁垒。另外两个比较有意思的方向,是(1)我们能不能把 GPT 这一套预训练的方法用在分子或者材料领域,来做一个材料大模型或者化学大模型,从而去降低或者减弱化学材料领域里获得实验数据比较昂贵的问题。(2)我们能不能转变材料设计的思维方式,从之前的筛选到我们现在用 GPT 这样的方法来生成一个新的材料。我觉得这两个方向上的探索都会比 GPT 直接的应用会更加令人兴奋一些。
贾皓钧:我稍微补充一点。首先 GPT 是大语言模型,如果你用 ChatGPT 频率高的话,就发现它的准确性是相对比较差的。但是科学问题,尤其是化学材料这种定量学科,非常注重精确。第二个问题,就是大语言模型目前很难生成出超出人类认知边界的内容。但做科研,一般来说我们就是要拓宽人类认知边界,发现新的现象和建立新的理论。第三个问题,做某一个垂直领域的大语言模型,比如材料、化学或物理之类的,能找到所有论文和公开数据库,一般都是已经做出来的东西,只有成功的案列,这个会导致训练集有很大的偏差。但我觉得之后各个领域,科研领域或学术领域,或某一个工业领域肯定会有垂直大模型出现。
ScienceAI:目前的 big tech(大型科技企业)对于 AI4S 有什么想法和行动?传统的材料化学制造业巨头是怎么看 AI4S?
贾皓钧:DeepMind 最早的 AlphaFold 算是 AI4S 出圈的一个产品。国外的tech 巨头都在 AI4S 投入非常多的真金白银,微软 2022 年开始专门成立了 AI4S 研究院,Meta 的 Open Catalysts 和 ESMFold,以及 Nvidia 最近的BioNeMo。在国内的话,字节跳动也已经有 AI4S 部门,以及深势科技这种专一的平台初创企业。AI4S 可以做的东西很多,也比较受资本的青睐。两个多月前,Meta 刚把 ESMFold 整个团队解散,但这个团队的 8 个核心成员两周后又融资到 40 million 美元,开始做他们模型的应用。大厂的逻辑,是背靠自己的算力和云服务,做一个平台,旨在把 AI4S 中比较成熟的方法产品化,以一些 ToB 或 ToC 的服务提供给大家。
从这个角度来看,传统的化工巨头其实是这些big tech 平台的用户。比如在微软最新推出的产品 Azure Quantum Elements(
https://quantum.microsoft.com/en-us/our-story/quantum-elements-overview),已经争取到了很多化学化工和材料的巨头,比如BASF,AspenTech ,Johnson Matthey。但同时这些化学材料厂内部也会有自己的数字创新团队以及 AI4S 方面的研究。但由于发展的历史路径不同,化学材料厂的的固定盈利模式都不太利于从原本的偏实验到偏 AI 的材料范式迁移的发生。
目前来看,不管是 big tech 还是传统的化学材料制造巨头都对 AI4S 非常感兴趣,也都花了非常多的金钱投入。但两边还是有一定的知识差,这需要花不少的时间去磨合,达成一致。
段辰儒:皓钧主要是从 big tech 角度说的,我从传统的材料化学制造业来聊聊。例如传统的材料化学制造业巨头:BASF,DOW,3M 等,他们正在努力将 AI 技术结合到其已有的工业生产技术上。从前端的材料的 R&D 产业到中游的生产反应条件优化。他们一般目前叫「数字化创新」(Digital Innovation),这个事情在产业内是达成共识的。具体投资情况,仅陶氏化学内部投入 Digital Innovation 去年是 4 亿美元,今年是 5.7 亿美元,明年会更多。除此之外,另外一个更重要的外部目标 2050 Carbon neutrality 的目标,优化和提升现有生产过程中的原料利用率和生产效率极为重要,尤其在目标开始初始阶段。
科技巨头和传统制造业巨头双方其实对 AI4S 都达成了一致,但是双方的出发点有不一样。这个事情就像自上而下(top down) 和自下而上(bottom up)的区别。举个例子,Microsoft 和 Google 它原本不是做这个行业的传统出身,那他们更倾向于从一个 top down 出发,从一个更高角度来提出一些模型来解决这个问题。这些传统的行业,可能是先优化现在的东西,例如材料配方或者反应条件,从 bottom up 来做。传统行业大规模用 AI4S 角度来做一些事情会很困难,因为其原本的研发路径和庞大的产业结构拖慢其 AI 变革的速度,船大掉头难。
ScienceAI:提到 AI 赋能企业科学研究,其中 AI 制药似乎占比最多?「AI+化学/材料」的产业落地更难吗?
贾皓钧:目前我个人感觉,就落地上来说,AI+化学/材料相对来说是容易的。其实我们组毕业的师兄师姐很多去了大药厂,而且剑桥就是生物医药的中心,其实我最早也考虑过做制药,但为什么没有做?
首先,药物从一个新化合物从最初的发现到申请上市,大约需要经过 15 年的时间。这个里面时间上最大的瓶颈是 clinical trail 的时间,而不是找药物分子的这个过程。但材料/化学发现,虽然也有后期的工程放大,但是目前核心的瓶颈还是找到一个合适的材料或者说是合适的配方。
另外,对于筛选的技术角度来说,一个核心的目标是从一个大的目标数据集,缩小到小的数据集,无论是小分子相互作用还是蛋白-蛋白相互作用,即使找到了合适的特定靶向药物,在人体复杂的环境中,也很难保证不进行其他的化学反应,药物递送本身就是一个很困难的问题。
最后,对于一个材料来说,能用不能用,从实验室,小试,中试,大试测一遍就知道。所以从这个角度来说,材料就更容易一些,也更能解决行业内的痛点。
段辰儒:我觉得这需要结合具体的情况和产业链,从工业的角度上来说,AI+化学/材料的优势是工业上扩大生产的能力比较成熟和标准化,尤其是国内化学化工产业的扩大生产能力其实非常强。我们主要的瓶颈反而是一开始怎么样能够发现一个更好的催化材料。在化学材料领域现在比较难的那部分是一开始的创新,而在药物领域其实有点相反,是最后面的临床试验、FDA 批准更加困难。所以,从逻辑上来讲,把 AI4S 应用在化学材料上做产业化反而会相对更加合理一些。
ScienceAI:从「基础科学研究」走向「工业落地」似乎一直是个难题,在 AI 助力的下,如何更好地加速产业落地?
段辰儒:落地的确是非常难的问题,因为单一模型的绝对领先既不是「工业落地」的充分条件,也不是必要条件。AlphaFold 作为 AI4S 领域最有代表性的工作之一,已经慢慢被应用在一些药物研发过程中。大家最近发现,虽然AlphaFold 可以产生非常好的蛋白质结构,但在 docking(即蛋白质和小分子药物的结合结构)预测上不尽如人意。很多人觉得这个结果出乎意料,而我觉得蛮正常的—— 因为 AlphaFold 在设计时就没有考虑到 docking。我们科学研究中遇到的问题,首先它是一个简化的问题,二它是一个局部的问题。简化的问题是指我们把问题简单化,做了一些近似。局部的问题,是指我们把一个非常长的产业链掏出来一部分,做一个问题。那在这种情况下,即使我们做的模型的准确率是 100%,也是没有办法「落地」的,因为它「既近似又片面」。
所以我觉得「落地」的难点,更多是对工业场景的了解和整合。比如,我们怎么样从 AI 的角度出发,把这些模型用到工业界当中,不断测试迭代,在工业界中形成一个比较好的工作流来切实解决问题。这其实是用更加发展的眼光看问题,相比于用 AI 的方法解决一个具体的问题,我们更需要以 AI 的角度持续思考和改进已有的工作流,即同时改进 AI 模型和问题本身。尤其是在现在这种开源的大背景之下,单一模型上的领先并不能给出非常大的壁垒。ChatGPT 一开始刚出来的时候,是「吊打」其他所有大模型的存在。但是经过一年的时间,在开源环境的不断促进下,大家和 ChatGPT 的差距也越来越小。所以,我觉得在「落地」角度,我们不应该只追求单一模型的准确,更多考虑如何根据实际问题研发出来有效的工作流,把不同的模型串联起来,并且与工业界紧密结合,不断迭代。
贾皓钧:从 「基础科学研究'」走向 「工业落地」一直都是一个复杂的挑战。首先,我们需要明确,基础科学研究的目标就不是直接为工业应用提供解决方案。举个例子,在科技发展的标度上,成熟工业可能处于 100 的级别,而基础科学研究则是从 0 到 1 或 0 到 0.1 的过程。这种研究通常发生在学校或科研院所,且并不是每个项目都能取得成功,也并不是每一个研究方向都会立即产生工业应用。
但这并不意味着所有的基础科学成果都无法转化为工业应用,因为在高校或研究机构中,尽管大部分研究是从 0 到 0.1 或者 0 到 1 的过程,但一旦某项成果成功,它可能会对特定行业产生深远的影响。这种情况在国内可能受到中美产业发展阶段的不同的影响。在美国,很多 0.1 到 100 的发展和 1 到 100 的发展都由初创公司推动,而国内也正在逐渐发展 「产学研」模式。
段辰儒:我心目中的科学工作者的理想是做两件事情。第一个是了解物质世界,这是比较基础的学科(物理、化学等)会做的事情,相当于探索世界的底层运行规律。第二个是改造世界,那改造世界其实更偏向于我们刚说的「落地」,可能是工程系做的事情。AI4S 是可以两者都具备的,它既可以帮助一些底层规律的发现(即 Science of AI),也可以用在更加偏应用的方面,来帮助我们更好的改造世界。
ScienceAI:在 AI4S 新的科研范式下,对于传统化学/材料学科的研究者,从原来的舒适圈转向交叉研究过程会遇到什么困难,如何去解决它?给我们打算即将进入交叉领域研究的后辈有什么建议?
贾皓钧:任何人都不可能做到全才,能把一件事情做到很好就已经非常了不起了。从更广的角度来说,衡量我们能不能做到一件事情通常只用考虑三个维度。首先你有没有能力来做这件事情?以转专业为例,假如从人文类学科想转到物理或者化学相对就比较难,你可能要在短期内大量学习各种新知识,同时面对很高的压力。第二个维度是你是否对这东西感兴趣。以读博来讲,那你是不是至少五年的读博期间有足够的兴趣来支撑。第三个维度就是这个东西未来有没有前景。所以我们追求的事情就是这三个事情的交集,可以从这三个维度分别想。
段辰儒:AI4S 并不是让大家把两门学科学的一样好,达到 5:5 的状态。即使达到 5:5 的水平,但缺乏思考以及没有足够多的上手经验依然很难有关键成就。我个人认为比较理想的是 3:7 或者 2:8 的状态。AI4S 的生态非常开放,有很多开源的代码库和开源工具大家可以去运用,同时也鼓励不同思想的碰撞。关键还是大家要有自己真正擅长的领域,之后主动去交流,其实并不用愁找不到互补的合作者。比如之前说的生成式 AI 做化学反应的工作,就是在一位有扩散模型经验的计算系小伙伴的帮助下合作的结果。
作为传统化学研究者,其实只需要花自己平时 20% 的时间去学习一些 AI 相关的知识,保证在和计算系的伙伴交流的时候,能够听懂对方的语言就已经足够了。这也是我们持续组织 AI4S workshop 的原因:提供一个交流的平台,让不同领域的人有机会都参加讨论自己的想法,壮大 AI4S 社区。
文章来自微信公众号 “ 机器之心pro ”,作者 MIT 贾皓钧&段辰儒博士


【开源免费】字节工作流产品扣子两大核心业务:Coze Studio(扣子开发平台)和 Coze Loop(扣子罗盘)全面开源,而且采用的是 Apache 2.0 许可证,支持商用!
项目地址:https://github.com/coze-dev/coze-studio
【开源免费】n8n是一个可以自定义工作流的AI项目,它提供了200个工作节点来帮助用户实现工作流的编排。
项目地址:https://github.com/n8n-io/n8n
在线使用:https://n8n.io/(付费)
【开源免费】DB-GPT是一个AI原生数据应用开发框架,它提供开发多模型管理(SMMF)、Text2SQL效果优化、RAG框架以及优化、Multi-Agents框架协作、AWEL(智能体工作流编排)等多种技术能力,让围绕数据库构建大模型应用更简单、更方便。
项目地址:https://github.com/eosphoros-ai/DB-GPT?tab=readme-ov-file
【开源免费】VectorVein是一个不需要任何编程基础,任何人都能用的AI工作流编辑工具。你可以将复杂的工作分解成多个步骤,并通过VectorVein固定并让AI依次完成。VectorVein是字节coze的平替产品。
项目地址:https://github.com/AndersonBY/vector-vein?tab=readme-ov-file
在线使用:https://vectorvein.ai/(付费)
【开源免费】DeepBI是一款AI原生的数据分析平台。DeepBI充分利用大语言模型的能力来探索、查询、可视化和共享来自任何数据源的数据。用户可以使用DeepBI洞察数据并做出数据驱动的决策。
项目地址:https://github.com/DeepInsight-AI/DeepBI?tab=readme-ov-file
本地安装:https://www.deepbi.com/
【开源免费】airda(Air Data Agent)是面向数据分析的AI智能体,能够理解数据开发和数据分析需求、根据用户需要让数据可视化。
项目地址:https://github.com/hitsz-ids/airda
【开源免费】XTuner 是一个高效、灵活、全能的轻量化大模型微调工具库。它帮助开发者提供一个简单易用的平台,可以对大语言模型(LLM)和多模态图文模型(VLM)进行预训练和轻量级微调。XTuner 支持多种微调算法,如 QLoRA、LoRA 和全量参数微调。
项目地址:https://github.com/InternLM/xtuner