# 热门搜索 #
大模型
人工智能
openai
融资
chatGPT
用扩散模型预测化学反应,速度直接暴涨1000倍!
原本需要用计算机硬算几小时甚至一天,现在单个GPU用6秒钟就能搞定。
这是MIT和康奈尔大学联合搞出来的一项新研究,用扩散模型来预测化学反应中最关键的过渡态结构,不仅计算速度提升1000倍,结果也竟然意想不到得准确,相关研究工作发表在Nature Computational Science上:

其中,麻省理工学院的段辰儒博士是第一加通讯作者。此外,康奈尔大学博士生杜沅岂、麻省理工学院博士生贾皓钧以及麻省理工学院Heather Kulik教授为该论文的共同作者,目前研究已经被MIT News报道。

要知道,预测反应中的过渡态结构远非想象中简单——
由于能量较高,它存在的时间往往只有飞秒级,即秒的千万亿分之一。
因此,目前还没有一台实验设备能直接观察它,只能通过量子化学计算的方式来预测,计算量少则几小时,多则数天。

如今用扩散模型就能得到几乎一样的预测结果,这究竟是怎么做到的?
进一步地,用AI预测化学反应,又究竟可以被应用在哪些领域、起到哪些作用?
我们和论文的其中两位作者,来自MIT的段辰儒和康奈尔大学的杜沅岂聊了聊,探讨了一下这项研究具体的原理、背后潜在的应用方向以及关于AI for Science的一些思考。
首先,需要理解为啥过渡态是研究化学反应的关键。
自远古的“炼金术”以来,化学一直是一门了解和控制物质之间相互作用的学科,化学反应又是其中非常核心的概念。

通常来说,一个化学反应由三个东西组成:反应物、生成物和过渡态结构。
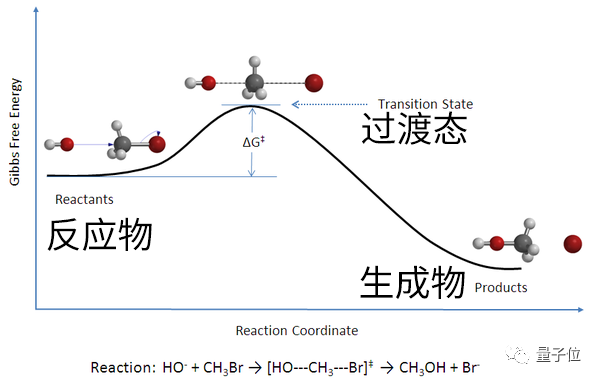
反应物和生成物大家都很熟悉了(如氢氧生成水),但实际上,二者之间的过渡态才是解释化学反应的关键——
作为化学反应过程中能量最高的状态,过渡态既能用来理解化学反应的机理、也能估算反应速率和能量。
一言以蔽之,要想真正搞懂、设计、优化并调控一个化学反应,就必须从过渡态结构下手。
所以,过渡态结构究竟要如何研究,又为什么需要用AI来做预测?
研究过渡态结构,不能只用分子结构来分析,而要更进一步研究它的3D构象。
分子结构,指为了便于理解化学反应,会人为构造出单双键这样的概念,并用键合关系、原子种类和数量来表示化学反应。
但要想真正精确计算化学反应的速率和能量,就必须要研究原子在3D空间中的位置关系(用3D坐标表示),即3D构象。只有对比原子在3D空间中的位置关系,才能进行量化分析,从而预测反应发生的过程。
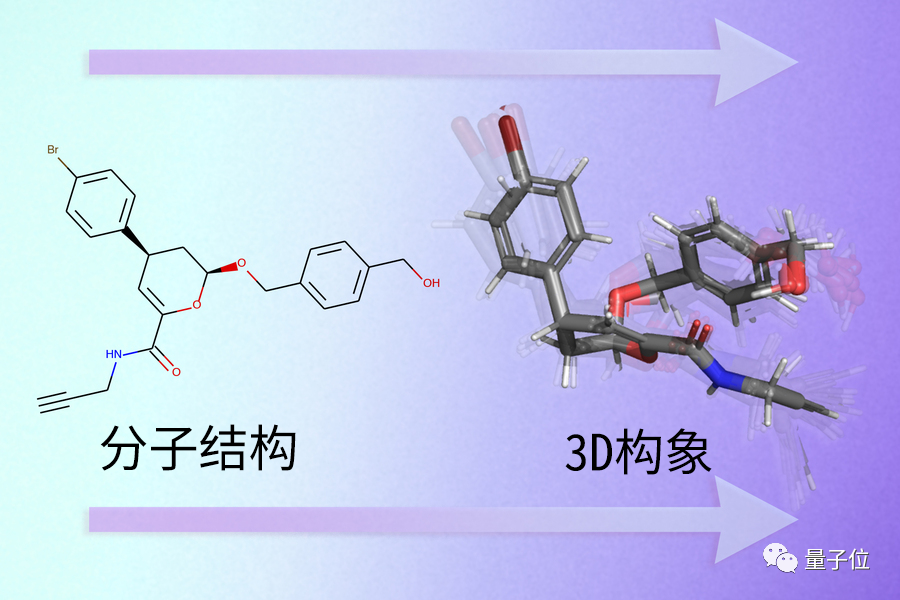
然而,相比反应物和生成物,过渡态存在的时间非常短,甚至只有飞秒量级。
受限于实验设备精度,过渡态结构无法用肉眼直接观察,此前只能通过量子力学方法——薛定谔方程来计算。
薛定谔方程,量子力学中的基本方程,描述粒子在某段时间内的状态如何变化。
BUT,手搓薛定谔方程计算量巨大,一个苯环的化学反应可能都需要算上一周时间,尤其随着体系增大(原子数量变多等),计算量更是呈现出指数级增长的趋势。
虽然后来出现了密度泛函理论(DFT)等近似方法加速计算,但算起来还是很慢、有时候甚至因为收敛性的问题导致计算不出来,“成本很昂贵,出错率还高”。

为此,来自MIT和康奈尔大学的研究人员,想到用AI的方法来直接预测过渡态结构,以节省计算量。
之所以选用扩散模型,据杜沅岂介绍,主要有三点原因:
其一,生成效果好。相比GAN,VAE等模型,扩散模型算是生成模型中效果较好的架构。
其二,更适合3D构象。相比分子结构是离散的数据,3D构象是一个连续的数据,且结构上涉及(平移、旋转和置换)对称性。
对于离散数据而言,基于机器学习+搜索的模型会比现有的(离散)扩散模型效果更好;但对于3D构象而言,无论是其对称性还是用连续数据表示,反而更适合用扩散模型生成。
其三,扩散模型自身的灵活性,使得它可以在生成时,保留一部分原有设计、填补剩余的部分,也使得它对于设计新的化学反应更有帮助。
具体到架构设计上,作者们将等变GNN融入到扩散模型中,设计了一种名叫OA-ReactDiff的结构,其中等变GNN能很好地处理并保留数据的对称性。
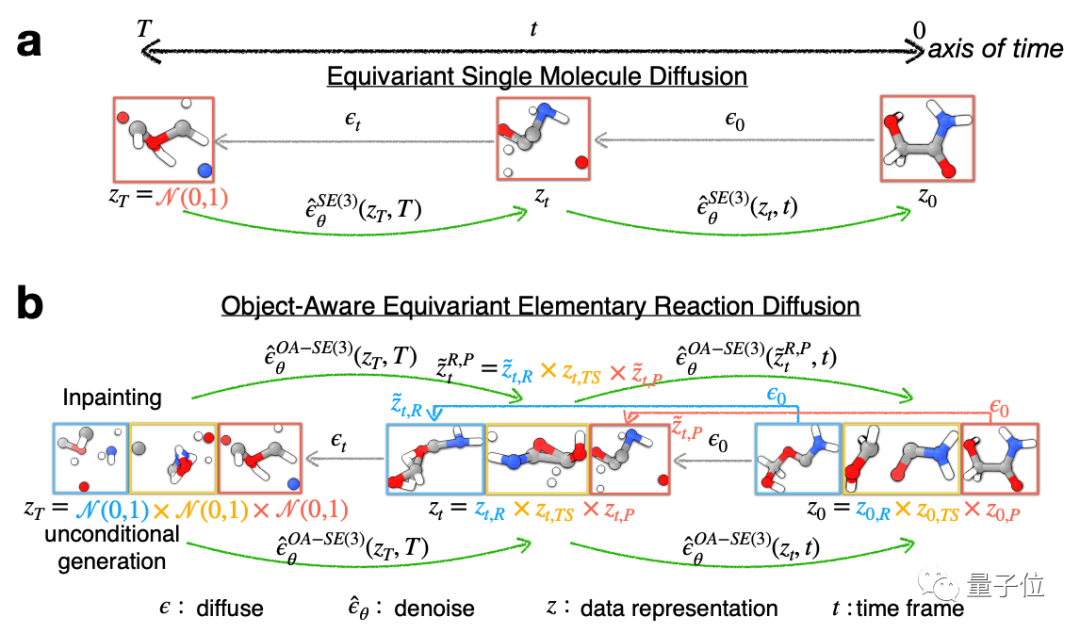
所以,OA-ReactDiff的效果究竟如何,或者说相比其他AI最大的亮点究竟是?
段辰儒表示,这项研究最核心的亮点主要有两个。
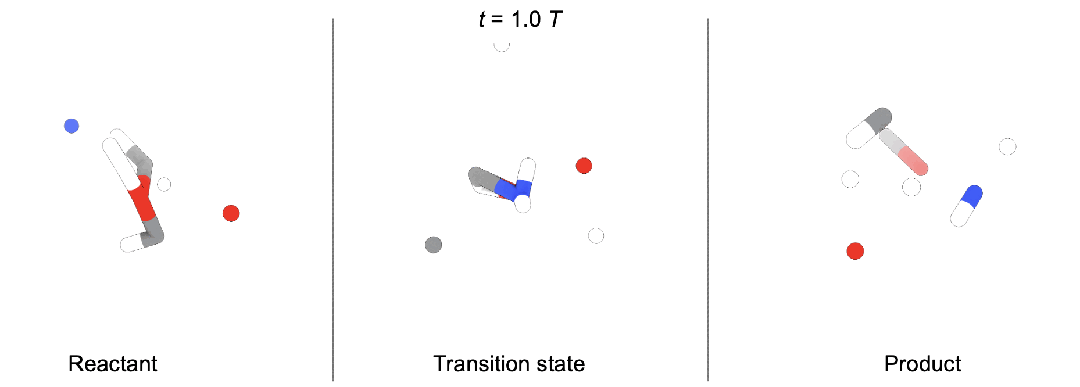
一个是预测准确性上,OA-ReactDiff的准确性超过了之前的一系列AI模型。
作者们选用了Transition1x作为数据集,这个数据集包含10073个化学反应,每个化学反应分别包含反应物、生成物和经过量子化学计算的过渡态结构,整个体系不超过23个原子。
这其中的9000个化学反应用作模型训练,1073个作为测试集,最终实现了0.183Å(1Å=0.1纳米)的平均均方根偏差和0.076Å的中位数均方根偏差。
相比于其他机器学习方法如PSI-based和NeuralNEB,OA-ReactDiff准确性都要更高。
与此同时,在没有刻意训练的情况下,OA-ReactDiff在更大的体系上也表现出了良好的预测能力。
另一个是预测速度上,OA-ReactDiff相比现有的计算方法,至少能快上1000倍。
这是因为,原本密度泛函理论算法的时间复杂度在O(N³)级别,但现在基于OA-ReactDiff的方法,直接将时间复杂度降低到O(N²)。
反映到现有数据集上,原本需要计算几小时甚至一天的化学反应,现在用OA-ReactDiff,只需要6秒钟就可以搞定。
进一步地,体系越大的化学反应,用扩散模型做预测的提速效果还会越好。
不过,这项研究究竟能被用在哪些地方呢?
我们也问了问两位作者关于这项研究潜在的应用、以及对于“用AI搞科学研究”这件事本身的看法。
对于研究本身,段辰儒认为主要有三个方面的作用。
最直观的一方面,自然是催化剂的设计了。
这也是研究人员接下来想要继续探索的方向,即继续扩展模型本身,加入催化剂等条件,并用AI来预测过渡态。
而在研究催化剂的基础上,又能进一步辅助燃料设计和新药开发。
以燃料为例,虽然涉及的燃烧反应体系往往不大,但发生反应的过程却极易被环境和条件影响。
因此,如果用AI来辅助预测化学反应的过程,或许能更快基于不同燃料发生反应的环境条件来预测效果。
这样一来,对于能源行业甚至航天行业也会产生进一步的影响。

最后,就是模拟自然界化学反应,来帮助开发新的理论研究。
像是模拟地球生命早期可能发生的反应、或是探索行星气体之间的相互作用等,通过用AI预测过渡态,也可能更快发现或验证新的理论,探究生命起源的意义。
听起来,AI似乎已经给化学研究带来了不少新的突破。
结合这段时间AI在生物制药(AlphaFold2)、以及材料学(GNoME)上取得的种种突破,对于生化环材这一系列理论学科而言,AI是否已经成为了新的技术主流路线?
更激进一点来说,对化学本身而言,未来AI是否会取代一部分现有的量子化学研究?
对此,两位作者均持有相同的观点,即AI(至少就监督模型而言),现阶段对于这些学科的影响力依旧是一种工具,而并非一条全新的技术路径。
这是因为,目前生化环材任何一个学科借助AI辅助研究的前提,依旧是已经有了一定的理论,而AI作为工具起到的是“锦上添花”的作用。
换言之,AI与这些学科的其他研究方法依旧是共存关系,并不会出现“替代”的情况。段辰儒对此做了一个有意思的比喻:
如果化学学科好比一个夹馍,机器学习方法和各种现有的化学方法,就是不同的食材。那夹馍里面可以夹肉,也可以夹其他的菜。
以近期大火的Google Deepmind研究GNoME为例,就是一个很好的“AI当工具人”的例子。
杜沅岂认为,如果从ML专业的角度去看待GNoME中涉及的AI方法,会发现它其实并不是最新的东西,像是其中涉及的几何深度学习,已经是这几年比较流行的研究方向。
但如果从材料学科的角度去看,这样大规模、高通量的稳定材料搜索,肯定还是有相当的科学意义在其中:
AI for Science本身,其实更侧重的是“Scientific Discovery”,它更大的意义在于科学发现。
而段辰儒认为,从应用的角度而言,无论是材料发现,还是预测化学反应,最终用AI去加速落地的宗旨依旧是“有用”二字:
正如这届NeurIPS AI4Science的workshop主题一样,“从理论到实践“。我认为这是AI4Science后面十年的必经之路,也是AI4Science的中期使命。
像OA-ReactDiff接下来的目标是找到一个可以应用的路径一样,GNoME发现220万稳定材料也只是应用的一个起点,接下来还需要很多的标准(如导热、导电等),才能让化学和材料真正在某一行业“发光发热”。
文章链接:C. Duan*, Y. Du, H. Jia, and H. J. Kulik, “Accurate transition state generation with an object-aware equivariant elementary reaction diffusion model”, Nat. Comput. Sci., ASAP, https://rdcu.be/dtGSF; https://www.nature.com/articles/s43588-023-00563-7


