# 热门搜索 #
大模型
人工智能
openai
融资
chatGPT
历时四年,微软亚研院AI for Science团队发布AI驱动的从头算(ab initio)生物分子动力学模拟系统。
直接登上Nature正刊。
系统名为AI²BMD,能够高效模拟含有10000多个原子的各种蛋白质,分辨率达到全原子级别,近似达到从头算(第一性原理)的精确度。
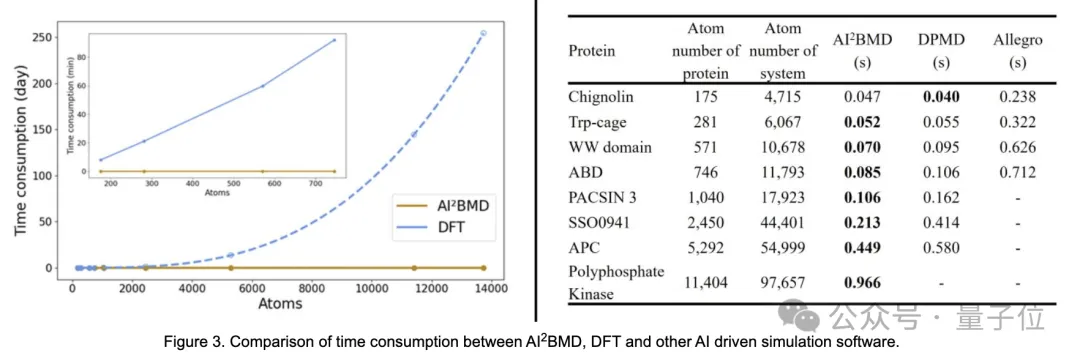
且比量子力学方法中的密度泛函理论(DFT)能实现的速度快几个数量级。

AI²BMD的出现,可促进药物发现、蛋白质设计和酶工程等领域的新生物医学研究。
微软研究院院长Peter Lee亲自发帖子为自家研究做宣传:
网友们纷纷表示这项研究十分令人着迷,可能会对一些领域产生颠覆性的影响:
可能会彻底改变我们对生物学和药物发现的认知。
为便于大家理解,作者们还自撰了一篇Blog,量子位在不改变原意的基础上进行了编译整理,一起来康康AI²BMD到底长啥样。

在介绍AI²BMD之前,先来简单了解一下分子动力学模拟。
生物世界的本质在于其分子及其相互作用的不断变化。理解生物分子的动态和相互作用对于破译生物过程背后的机制以及开发生物材料和药物至关重要。然而通过实验几乎不可能捕捉这些现实生活中的运动。
此前,AlphaFold、RoseTTAFold等为代表的深度学习方法,已经实现了用实验精度预测静态晶体蛋白结构。
不过精确地在原子分辨率下表征动态仍然是一项非常具有挑战性的任务,特别是当蛋白质发挥作用并与其它生物分子或药物分子相互作用时。
分子动力学(MD)模拟方法将物理学定律与数值模拟相结合,用于探索分子运动与其生物功能之间的关系,已经被广泛使用了几十年。
分子动力学模拟大致可以分为两类:经典分子动力学和量子力学。
经典版本在2013年获得诺贝尔奖,强调了其在推动人类理解复杂生物系统方面的关键作用。

量子力学方法中的密度泛函理论(DFT)在1998年获得诺贝尔奖,标志着计算化学的一个关键时刻。

不过,经典分子动力学采用分子系统的简化表示,实现长时间构象变化的快速模拟速度,但准确性较低。相比之下,像密度泛函理论这样的量子力学模型提供了自下而上的计算,但对于大生物分子来说,计算成本过高。
由此,微软研究院开发出了基于AI的从头算生物分子动力学系统——AI²BMD,这也是团队四年研究成果的结晶。
AI²BMD能够高效地以全原子分辨率模拟包含超过10000个原子的各类蛋白质,达到近似从头计算(第一性原理)的精度。
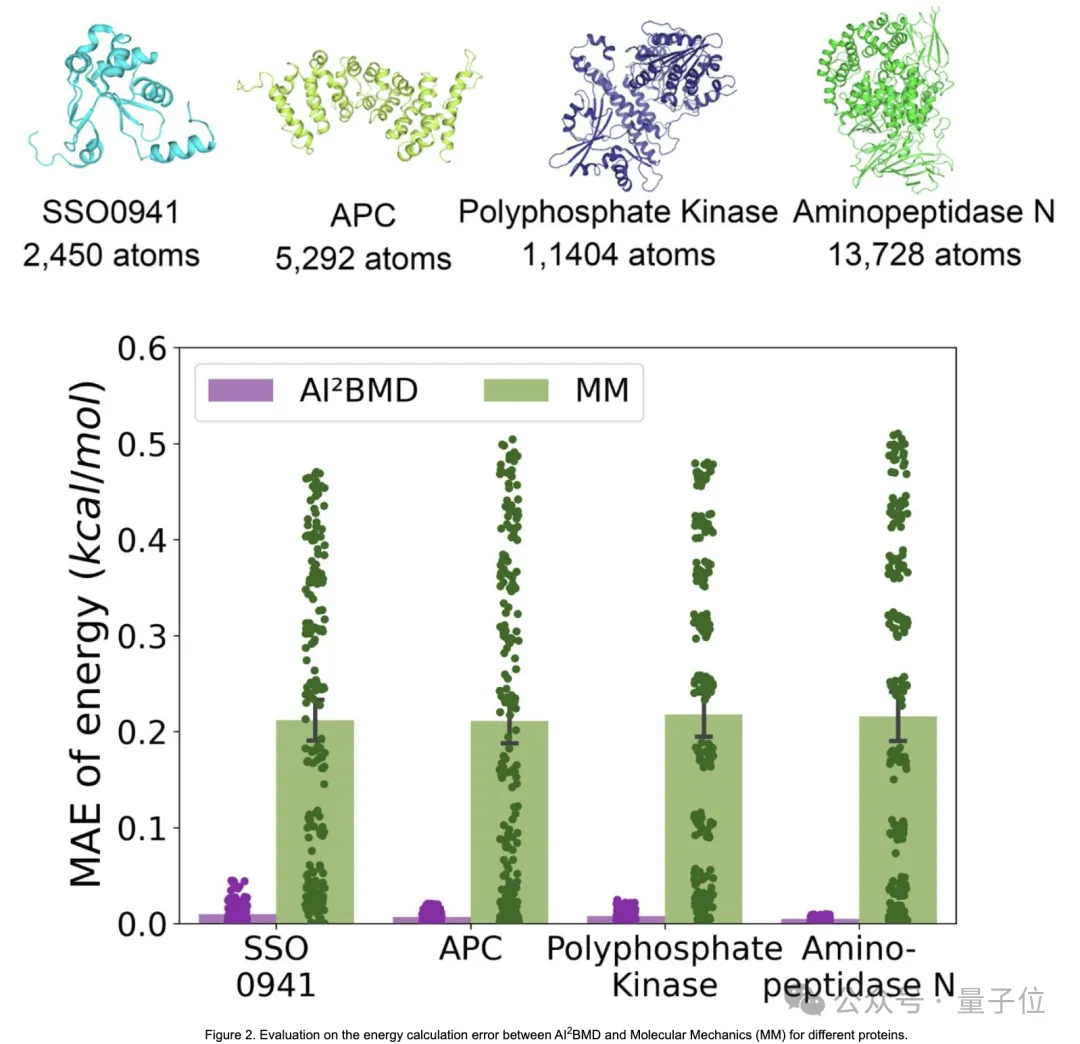
相比标准模拟技术,它在生物分子模拟方面实现了前所未有的权衡:获得比经典模拟更高的精度,其计算成本虽然高于经典模拟,但比密度泛函理论(DFT)快几个数量级。

AI²BMD采用专门设计的通用蛋白质分片方法,将蛋白质分割成重叠的单元,创建了2000万个快照的数据集,这也是迄今为止DFT层面最大的数据集。
基于团队先前设计的通用分子几何建模基础模型ViSNet,团队用机器学习训练了AI²BMD的势能函数。然后通过高效的AI²BMD模拟系统进行模拟,在每一步中基于ViSNet的AI²BMD势能都会以从头计算的精度计算蛋白质的能量和原子力。

通过动力学和热力学的全面分析,AI²BMD与实验室数据表现出更好的一致性,例如在蛋白质的折叠自由能和不同现象方面,比经典分子动力学表现更好。
总结来说,AI²BMD在分子动力学模拟领域取得了以下几个方面的重大突破:
引入了一种可泛化的“机器学习力场”,这是一种学习原子与分子间相互作用的模型,用于具有从头算精度的全原子蛋白质动力学模拟。
它是首个解决机器学习力场在模拟蛋白质动力学中的泛化挑战的方法,展示了对多种蛋白质进行稳健地从头算分子动力学模拟。
AI²BMD将量子力学建模从小的局部区域扩展到整个蛋白质,而且不需要对蛋白质有任何先验知识。这消除了量子力学与蛋白质的分子力学计算之间可能的不兼容性,并将量子力学区域的计算速度提高了几个数量级,使全原子蛋白质接近从头算计算成为现实。因此,AI²BMD为众多下游应用铺平了道路,提供了表征复杂生物分子动力学的新视角。
AI²BMD比x和其它量子力学方法快几个数量级。支持对超过10000个原子的蛋白质进行从头计算,使其成为跨学科领域中最快的AI驱动的分子动力学模拟程序之一。
对于AI²BMD和蛋白质分子力学模拟的蛋白质折叠和解折叠过程,AI²BMD能够探索更多蛋白质分子力学无法检测到的可能构象空间。
因此,AI²BMD为研究药物-靶标结合过程、酶催化、变构调节、内源性无序蛋白等过程中的蛋白质柔性运动提供了更多机会,与湿实验更好地吻合,为生物机制检测和药物发现提供更全面的解释和指导。

AI²BMD超越了量子力学/蛋白质分子力学混合方法,并在不同的生物应用场景中展示了与湿实验室实验的高度一致性,包括J耦合、焓、热容、折叠自由能、熔点和pKa计算。
这项研究共同一作有四位Tong Wang、Xinheng He、Mingyu Li、Yatao Li,由Tong Wang、Bin Shao共同指导。
Tong Wang

Tong Wang是微软研究院AI4Science高级研究员。他在清华大学获得博士学位,并在哈佛大学有访问学者经历。
他的研究重点是分子动力学模拟、量子模拟、计算机辅助药物发现和蛋白质结构预测的算法设计与应用。
Tong Wang博士以第一作者和通讯作者身份发表了一系列研究,刊登在Nature Machine Intelligence、Nature Communications、Cell Research等期刊上,并拥有多项专利。他还是Nature系列期刊的审稿人以及ACS出版物的荣誉审稿人。
作为团队负责人,他在首届全球AI药物开发大赛中获得冠军,并在NIPS2022的OGB大规模挑战赛中获胜。
邵斌(Bin Shao)

邵斌是微软研究院AI4Science资深高级研究经理,于2010年7月在复旦大学获得博士学位后加入微软。
他的研究兴趣包括蛋白质动力学模拟、计算生物学、机器学习和分布式计算,研究成果已发表在多个顶会和期刊上。
由邵斌团队开发的Microsoft Graph Engine正在为微软的多个产品和服务提供支持,如微软Satori知识图谱、必应搜索、MSN、Xbox和认知服务等。邵斌还是开源轻量级AIMD模拟程序LightAIMD的发起人和主要开发者。
参考链接:
[1]https://www.nature.com/articles/s41586-024-08127-z
[2]https://x.com/peteratmsr/status/1854199356850889096
[3]https://www.binshao.info/
[4]https://www.microsoft.com/en-us/research/people/watong/
文章来自于微信公众号 “量子位”,作者“西风”



【开源免费】DeepBI是一款AI原生的数据分析平台。DeepBI充分利用大语言模型的能力来探索、查询、可视化和共享来自任何数据源的数据。用户可以使用DeepBI洞察数据并做出数据驱动的决策。
项目地址:https://github.com/DeepInsight-AI/DeepBI?tab=readme-ov-file
本地安装:https://www.deepbi.com/
【开源免费】airda(Air Data Agent)是面向数据分析的AI智能体,能够理解数据开发和数据分析需求、根据用户需要让数据可视化。
项目地址:https://github.com/hitsz-ids/airda
